
How to judge whether QSAR/read-across predictions can be trusted: a novel approach for establishing a model's applicability domain†

Abstract
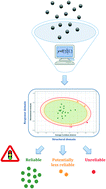
The EU REACH legislation, the OECD and US EPA official guidance documents, as well as the 3Rs principle (replacement, reduction, refinement of animal testing), all advocate the necessity of developing comprehensive computational methods (e.g. quantitative structure–activity relationship, read-across) that would enable the predictive modeling of both chemical (e.g. nanoparticle) specific functionalities and their hazards. However, since computational (nano)toxicology continues to ‘learn on the fly’ and relies on the use of a vast array of innovative machine-learning algorithms, serious concerns about the reliability of in silico predictions are raised. This study aimed to give an answer to the following question: how to judge whether QSAR/read-across predictions are reliable. Here, an effective approach for graphical assessment of the limits of a model's reliable predictions (so-called applicability domain, AD) was introduced. The probability-oriented distance-based approach (ADProbDist) was proposed as a robust and automatic method for defining the interpolation space where true and reliable predictions can be expected. Its usefulness was confirmed by using four nano-QSAR/read-across models recently reported in the literature. The results of the study showed that the ADProbDist approach is more restrictive in terms of the chemical space that falls in the AD of a model than the range, geometrical, distance and leverage approaches. The advantages of the proposed ADProbDist approach include (but are not limited to) the fact that it works with relatively small datasets and enables the identification of (un)reliable predictions for newly screened chemicals without experimental data. Further, to facilitate the use of the ADProbDist approach, this study provides the developed in-house R-codes.
 Please wait while we load your content...
Please wait while we load your content...

