
Band engineering of multicomponent semiconductors: a general theoretical model on the anion group†
 ‡*ab
G. W.
Qin
*b
‡*ab
G. W.
Qin
*b
Abstract
Development of energy conversion semiconductor materials has attracted increasing interest over the past three decades, but most successful semiconductors are unary or binary, rather than multicomponent semiconductors (MCSCs). There is a several orders of magnitude wider variety of MCSCs than unary and binary semiconductors, but very few electronic energy theories have been able to deal with more than two composition variables so far, and thus desired MCSCs are hard to predict. In this work, we propose a universal anion group model based on the analysis of electronic structures in an ABO3 perovskite prototype. Under a first order approximation, that is, the ‘A’ cation and the (BO6) anion group have very little hybridization, we find that the band gap of the ABO3 semiconductor is mainly determined by the (BO6) anion group and is very similar to that of binary compounds consisting of the same anion group constituents, while the band edges can be adjusted by the ‘A’-site cation. When more intense hybridizations exist, the predicted results can be amended by considering the higher order approximation. Using this model, the band gaps and edges of quaternary AgxNa(1−x)NbO3 perovskites and ZnxMg(1−x)Fe2O4 spinels have been predicted and are consistent with reported experiments and first principles calculations, further confirming the validity of the proposed model. Therefore, an anion group model on MCSCs can not only promote the probability of success in band engineering, but can also pave the way for speeding up the design of novel and desired MCSCs using known binary semiconductors for use in the field of energy conversion materials as photocatalysts, light-emitting materials, complementary light-absorption materials for solar cells, etc.
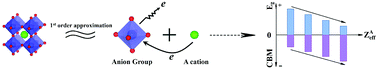
 Please wait while we load your content...
Please wait while we load your content...

