
Synthesis, characterization, and catalytic application in aldehyde hydrosilylation of half-sandwich nickel complexes bearing (κ1-C)- and hemilabile (κ2-C,S)-thioether-functionalised NHC ligands†

Abstract
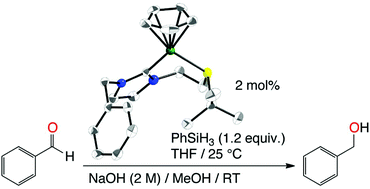
Neutral nickel–N-heterocyclic carbene complexes, (κ1-C)-[NiCpBr{R-NHC-(CH2)2SR′}] [Cp = η5-C5H5; R-NHC-(CH2)2SR′ = 1-mesityl-3-[2-(tert-butylthio)ethyl]- (1a), 1-mesityl-3-[2-(phenylthio)ethyl]- (1b), 1-benzyl-3-[2-(tert-butylthio)ethyl]- (1c), 1-benzyl-3-[2-(phenylthio)ethyl]-imidazol-2-ylidene (1d)], which bear a N-bound thioether side arm, were prepared by the reaction of nickelocene with the corresponding imidazolium bromides [R-NHC-(CH2)2SR′·HBr] (a–d), via conventional or microwave heating. The 1H NMR spectra of the benzyl-substituted species 1c and 1d showed signals for diastereotopic NCH2CH2S protons at room temperature. However, structural studies established the absence of coordination of the sulphur atom in the solid state, and solvent DFT calculations showed that bromide displacement by sulphur is an unfavourable process (ΔG = +13.5 kcal mol−1 for 1d), thereby suggesting that the observed disatereotopicity is more likely due to significant steric congestion rather than to a possible C,S-chelation in solution. Treatment of these complexes with KPF6 in tetrahydrofuran (THF) led to bromide abstraction to afford the cationic complexes [NiCp{R-NHC-(CH2)2SR′}](PF6) (2a–c). Alternatively, 2a–c could also be prepared by the direct reaction of nickelocene with the corresponding imidazolium hexafluorophosphate salts [R-NHC-(CH2)2SR′·HPF6]. Inversely to the neutral species, whereas X-ray crystallography established C,S-chelation in the solid state, the 1H NMR spectra (CDCl3, CD2Cl2, or thf-d8) at room temperature showed no diastereotopic NCH2CH2S protons, thus suggesting the possible displacement of the sulphur atom by the respective solvents and/or very fast sulphur inversion. DFT calculations established a low energy inversion process in all cases (+9 ≤ ΔG‡ ≤ +13 kcal mol−1) as well as a favourable solvent coordination process (ΔG‡ ≈ +11 kcal mol−1; ΔG ≈ −7 kcal mol−1) with a solvent such as THF, thus suggesting that sulphur inversion and/or solvent coordination can both account for the absence of diastereotopy at room temperature, depending on the solvent. While all complexes catalysed the hydrosilylation of benzaldehyde in the absence of any additive, the cationic C,S-chelated complexes 2 proved more active than the sterically constrained neutral species 1. In particular, 2c proved to be the most active pre-catalyst and its catalytic charge could be lowered down to 2 mol% with PhSiH3 as the hydrogen source.
 Please wait while we load your content...
Please wait while we load your content...

