
Slow spin crossover in bis-meridional Fe2+ complexes through spin-state auto-adaptive N6/N8 coordination†

Abstract
Fe2+ spin crossover (SCO) complexes with long-lived excited high-spin (HS) states are promising molecular switches. An enhanced kinetic stability of spin-state isomers can be expected to foster applications beyond the limits of cooperative SCO. In this study, we describe a new approach to slow down the spin-state exchange by simple commutation of a phenyl substituent by a pyridyl substituent. To this end, N4 ligand 6-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)-2,2′-bipyridine (3b) is synthesized as an N4 homologue of the well-established meridional N3 ligands motif. Phenyl-substituted 6-(3-phenyl-1H-pyrazol-1-yl)-2,2′-bipyridine (3a) serves as an intrinsic N3 reference throughout. 3b offers variable coordination numbers, N3 versus N3(+1) and N4, reflecting the preferences of the metal center. As is shown herein through an extended solid-state structure-chemical and solution-state NMR study, which is augmented by density-functional theory modeling, both the coordination geometry and its structural dynamics are indeed highly sensitive towards the expansion of the nominal donor number. The additional donors in 3b introduced through the phenyl-pyridine commutation actually give rise to a rich and diverse stereochemistry of the derived Zn2+ and Fe2+ complexes. Notably, even within a single complex unit coordination of 3b ranges from strongly distorted N3 coordination with a long assisting additional contact (Zn2+ and Fe2+) to a more symmetric N2(+2) or N4 situation in Fe2+. DFT modeling unravels that the additional donors are hemi-labile and coordinate to the Fe2+ only in HS state, leaving the elusive low-spin (LS) state in a fairly undisturbed octahedral environment with 3b being N3 coordinate. That is, the coordination number of the complex autogeneously responds to the altered spin-state. Necessarily this switch in coordination number requires strong structural changes upon SCO. This leads to increased activation barriers for SCO as could be deduced from a temperature-dependent analysis of the dynamic 1H NMR-line broadening and corroborated by accompanying theoretical analysis of the SCO reaction coordinate. For [Fe(3b)2]2+ long spin-state lifetimes τ > 1 ms prevail below the characteristic temperature T (1 ms) = 235 K; this value should be compared with a lifetime of only 150 ns derived for the close analogue [Fe(3a)2]2+. The principle applied herein is general and allows transferring of LS Fe2+ complexes with suitably placed phenyl substituents into SCO complexes with spin-state adaptive coordination number and hence long-lived HS excited states.
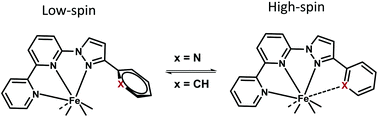
 Please wait while we load your content...
Please wait while we load your content...

