
Tuning the affinity of catechols and salicylic acids towards Al(iii): characterization of Al–chelator interactions†

Abstract
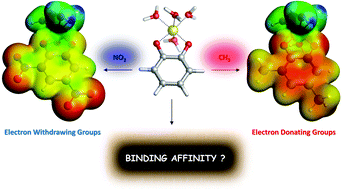
Due to aluminum's controversial role in neurotoxicity, the goal of chelation therapy, the removal of the toxic metal ion or attenuation of its toxicity by transforming it into less toxic compounds, has attracted considerable interest in the past years. In the present paper we present, validate and apply a state-of-the-art theoretical protocol suitable for the characterization of the interactions between a chelating agent and Al(III). In particular, we employ a cluster-continuum approach based on Density Functional Theory calculations to evaluate the binding affinity of aluminum for a set of two important families of aromatic chelators: salicylic acids and catechols. Our protocol shows very good qualitative agreement between the computed binding affinities and available experimental stability constants (log β) values for 1 : 1, 1 : 2 and 1 : 3 complexes. Then, we have investigated the nature of the Al–O bond in an enlarged dataset of 27 complexes of 1 : 1 stoichiometry, by means of the QTAIM and Energy Decomposition Analysis (EDA). Quite interestingly, we have found that although the Al–O interaction is mainly electrostatic, there is a small but significant degree of covalency that explains the modulation of binding affinities in both families of compounds by the addition of electron donating (CH3, OCH3) or withdrawing (NO2, CF3) substituents. The role of aromaticity and the mechanisms of action of the different functional groups were also evaluated. Finally, we have analyzed the competition between Al(III) and proton toward the binding of these chelators, giving a rationalization of the different trends found experimentally between log β and the amount of free aluminum in solution in the presence of a given ligand (p[Al]). In summary, we propose a validated and comprehensive computational protocol that can provide a valuable help toward the design and tuning of new efficient aluminum chelators.
 Please wait while we load your content...
Please wait while we load your content...

