
How the distribution of reduced vanadium centers affects structure and stability of the MoVOx material†

Abstract
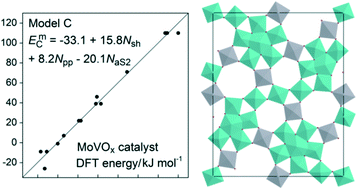
With hybrid density functional theory calculations, we examined how the locations of reduced V sites affects the stability of the bulk MoVOx catalyst material. For 14 exemplary distributions of reduced centers, V4+, the total energies varied by ∼140 kJ mol−1 per unit cell. Aiming at a conceptual understanding of the stability of this transition metal oxide material, we identified three factors that lead to low-energy configurations: (i) the type of reduced V centers; (ii) the number of notable polaron–polaron interactions, i.e., configurations where two V4+ centers are separated by a single intermediate Mo center; (iii) the number of pentameric units with a single V4+ center. Hence, for a more stable material, V centers in the linker units S1 and S3 should first be reduced, followed by a single S7 site per pentameric unit, and, in lower priority, reduced V centers in close proximity should be avoided. Based on this insight, we present a linear relation that predicts the stability of such configurations with a standard error of only 6 kJ mol−1. Accordingly, the pentameric units of low-energy variants of the MoVOx catalyst material are in a favorable configuration, as proposed for hydrocarbon oxidation.
 Please wait while we load your content...
Please wait while we load your content...

