
Ab initio quantum-chemical computations of the absorption cross sections of HgX2 and HgXY (X, Y = Cl, Br, and I): molecules of interest in the Earth's atmosphere†

Abstract
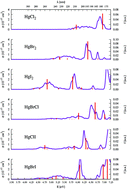
The electronic-structure properties of the low-lying electronic states and the absorption cross sections (σ(E)) of mercury halides HgCl2, HgBr2, HgI2, HgBrCl, HgClI, and HgBrI have been determined within the UV-vis spectrum range (170 nm ≤ λphoton ≤ 600 nm) by means of the DKH3-MS-CASPT2/SO-RASSI quantum-chemical methodology (with the ANO-RCC basis set) and a semi-classical computational strategy based on nuclear sampling for simulating the band shapes. Computed band energies show a good agreement with the available experimental data for HgX2 with errors around 0.1–0.2 eV; theoretical and σ(E) are within the same order of magnitude. For the mixed HgXY compounds, the present computed data allow us to interpret previously proposed absorption bands estimated from the spectra of the parent molecules HgX2 and HgY2, measured in methanol solution. The analyses performed on the excited-state electronic structure and its changes around the Franck–Condon region provide a rationale on the singlet–triplet mixing of the absorption bands and the heavy-atom effect of the Hg compounds. Furthermore, the present benchmark of HgX2 and HgXY absorption σ values together with the previous benchmark of the electronic-structure properties of HgBr2 [see S. P. Sitkiewicz, et al., J. Chem. Phys., 2016, 145, 244304] has been helpful to set up a methodological and computational protocol which shall be used for predicting the atmospheric absorption and photolysis properties of several Hg compounds present in the atmospheric cycle of Hg.
 Please wait while we load your content...
Please wait while we load your content...

