
Dynamics and kinetics of the reaction OH + H2S → H2O + SH on an accurate potential energy surface

Abstract
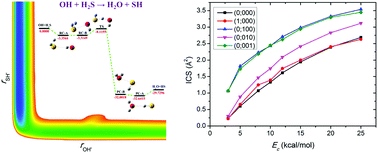
The dynamics and kinetics of the prototypical hydrogen abstraction reaction OH + H2S → H2O + SH were studied using the quasi-classical trajectory approach on a new accurate ab initio potential energy surface (PES) for the ground electronic state. The PES was developed by fitting 82 680 ab initio points at the level of UCCSD(T)-F12a/aug-cc-pVTZ using the fundamental invariant-neural network method. On one hand, excitation of either the symmetric stretching mode or the asymmetric stretching mode of the reactant H2S almost equivalently enhances the reaction. The promotional effect of exciting the bending mode of H2S is not as strong as exciting the stretching modes while it increases with the collision energy. On the other hand, the calculated vibrational state distribution of the product H2O based on the normal mode analysis method agrees reasonably well with the earlier experimental result, which was rationalized by the underlying reaction mechanisms. In addition, the rate constants of the reaction have a non-Arrhenius temperature dependence.
 Please wait while we load your content...
Please wait while we load your content...

