
Stability of AumAgn (m + n = 1–6) clusters supported on a F-center MgO(100) surface†
 a
and
Marcela R.
Beltrán
*a
a
and
Marcela R.
Beltrán
*a
Abstract
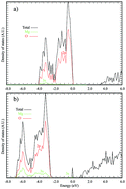
A theoretical study has been performed for deposited AumAgn (m + n = 1–6) clusters. The combined use of the Mexican Enhanced Genetic Algorithm (MEGA) and Density Functional Theory (DFT) calculations allows us to explore the potential energy surface and therefore, find the global minimum configuration for each composition. We have performed calculations of clusters deposited on defects (oxygen vacancies) known as F centers on MgO (100) surfaces. Our results show interesting differences in the geometries of the clusters upon deposition and as a consequence in their electronic properties. The combination of two metals with different electronegativities creates an inhomogeneous charge distribution on their exposed surface producing good conditions for a catalytic process to take place.
 Please wait while we load your content...
Please wait while we load your content...

