
Performance of TDDFT with and without spin-flip in trajectory surface hopping dynamics: cis–trans azobenzene photoisomerization†
 ab
Chaoyuan
Zhu
*ad
ab
Chaoyuan
Zhu
*ad
Abstract
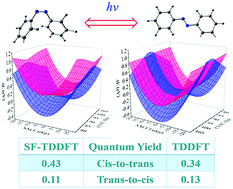
Time-dependent density functional theory (TDDFT) with and without a spin-flip scheme is extensively compared in on-the-fly trajectory surface hopping molecular dynamics with a global switching (GS) algorithm. The simulation is performed for cis–trans azobenzene photoisomerization following the excitation to the S1(nπ*) state that is involved in a conical intersection (CI) between ground and first excited states. This CI is found correctly to be a single-cone (artificial double-cone) structure computed by the TDDFT method with (and without) spin-flip. Nevertheless, simulated quantum yields and lifetimes are in very good agreement; 0.43 and 63 fs (0.34 and 62 fs) for cis-to-trans isomerization, and 0.11 and 2200 fs (0.13 and 1040 fs) for trans-to-cis isomerization, by TDDFT with (and without) a spin-flip scheme. Distributions of excited-state decay, hopping spots and products, as well as typical trajectories have similar patterns and behaviors with and without spin-flip. The global switching trajectory surface hopping method is demonstrated to be well suited to TDDFT on-the-fly dynamic simulation with and without spin-flip. For comparison, previous simulations with the CASSCF method and Tully's fewest-switches trajectory surface hopping method are also addressed.
 Please wait while we load your content...
Please wait while we load your content...

