
Probing the binding mode and unbinding mechanism of LSD1 inhibitors by combined computational methods†
 *a
*a
Abstract
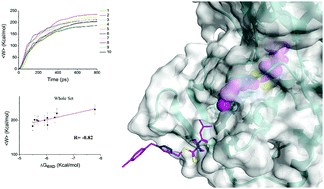
Lysine specific demethylase 1 (LSD1) has emerged as a potential drug target in cancer therapy and a variety of inhibitors have been reported. We have recently reported the discovery of a series of triazole-dithiocarbamate based compounds, which were basically confirmed as cofactor flavin adenine dinucleotide (FAD)-competing inhibitors by experiments. However, the binding modes of the inhibitors to the binding site were undetermined. Here, we employed computational methods including molecular docking, classical molecular dynamics (MD) and steered molecular dynamics (SMD) simulations to investigate the potential binding modes of these inhibitors to LSD1. Based on the high correlation between the mean non-equilibrium pulling work 〈W〉 and experimental binding affinity, we identified the optimal binding modes of this class of compounds with LSD1. Using the optimal inhibitor binding conformation, we then performed SMD to study the ligand unbinding mechanism with a lower pulling velocity at 0.0005 nm ps−1. We found that residue Arg316 plays a crucial role in the binding/unbinding process. Furthermore, a gatekeeper residue Trp756 influences the ligand unbinding process by acting like a switch via steric hindrance but can enhance the hydrophobic interaction with the inhibitor. Hydrophobic interaction also dominated the interaction between LSD1 and the inhibitors. The pivotal residues and interactions between LSD1 and inhibitors determined from this study can be used to improve the inhibition activity of this series of inhibitors in development and to discover new scaffolds as FAD-competing inhibitors in compound screening.
 Please wait while we load your content...
Please wait while we load your content...

