
Fitting electron density as a physically sound basis for the development of interatomic potentials of complex alloys†
 a
Sofía
Calero,
a
Javier
Segurado,
*bc
Francisco
Montero-Chacón,
*de
A. Rabdel
Ruiz-Salvador
*a
and
Said
Hamad
*a
a
Sofía
Calero,
a
Javier
Segurado,
*bc
Francisco
Montero-Chacón,
*de
A. Rabdel
Ruiz-Salvador
*a
and
Said
Hamad
*a
Abstract
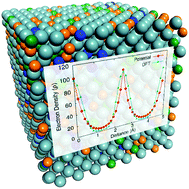
The development of new interatomic potentials to model metallic systems is a difficult task, due in part to the dependence between the parameters that describe the electron density and the short-range interactions. Parameter search methods are prone to false convergence. To solve this problem, we have developed a methodology for obtaining the electron density parameters independently of the short-range interactions, so that physically sound parameters can be obtained to describe the electron density, after which the short-range parameters can be fitted, thus reducing the complexity of the process and yielding better interatomic potentials. With the new method we can develop self-consistent, accurate force fields, using solely calculations, without the need to fit to experimental data. Density functional theory calculations are used to compute the observables with which the potential is fit. We applied the method to a Ni-based Inconel 625 superalloy (IN625), modelled here as Ni, Cr, Mo and Fe solid solution alloys. The capability of the force fields developed using this new method is validated, by comparing the structural and thermo-elastic properties predicted with the force fields, with the corresponding experimental data, both for single crystals and polycrystalline alloys.
 Please wait while we load your content...
Please wait while we load your content...

