
Lattice thermal conductivity of monolayer AsP from first-principles molecular dynamics†

Abstract
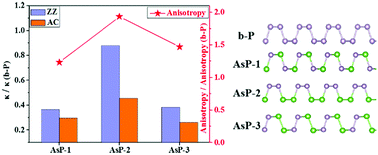
Few-layered arsenic–phosphorus alloys, AsxP(1−x), with a puckered structure have been recently synthesized and demonstrated with fully tunable band gaps and optical properties. It is predicted that the carrier mobility of monolayer AsP compounds is even higher than that of black phosphorene (b-P). The anisotropic and orthogonal electrical and thermal transport properties of the puckered group VA elements make them intriguing materials for thermoelectric applications. Herein, we investigated the thermal transport properties of AsP based on first-principles molecular dynamics and the Boltzmann transport equation. We reveal that monolayer AsP with three different chemical structures possesses thermal conductivities lower than b-P, but with increased anisotropy. Further, these structures behave profoundly different on heat conduction. This can be attributed to the distinct low-frequency optical modes associated with their bonding nature. Our results highlight the impact of atomic arrangement on the thermal conductivity of AsP, and the structure–property relationship established may guide the fabrication of thermoelectric materials via the engineered alloying method.
 Please wait while we load your content...
Please wait while we load your content...

