
Vibronic relaxation energies of acene-related molecules upon excitation or ionization†
 *a
and
*a
and
Abstract
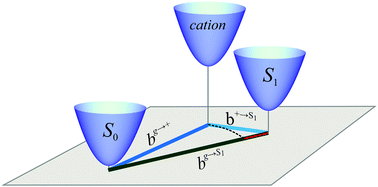
With the perspective of future vibronic studies, ab initio calculations of the energies of vibrational relaxation that follows the processes of singlet or triplet excitation, and positive or negative ionization, are reported for four series of compounds of potential interest in the context of photovoltaic applications, notably those rooted in singlet exciton fission. The commonly used method of evaluating the energy of vibrational relaxation following ionization of an excited molecule is examined and found to be dubious, especially when used within approaches based on the concept of an effective progression-forming mode. In this regard, methodological consistency in computing the relaxation energies and Franck–Condon parameters for different excitation processes is found to be of paramount importance. The presented results respond to the existing demand for vibronic coupling constants relevant to singlet fission.
 Please wait while we load your content...
Please wait while we load your content...

