
Phonons, phase transitions and thermal expansion in LiAlO2: an ab initio density functional study†

Abstract
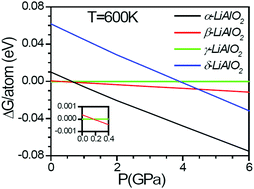
We have used the ab initio density functional theory technique to understand the phase transitions and structural changes in various high temperature/pressure phases of LiAlO2. The electronic band structure as well as phonon spectra is calculated for various phases as a function of pressure. The phonon entropy used for the calculations of Gibbs free energy is found to play an important role in the phase stability and phase transitions among various phases. A sudden increase in the polyhedral bond lengths (Li/Al–O) signifies the change from the tetrahedral to octahedral geometry at high-pressure phase transitions. The activation energy barrier for the high-pressure phase transitions is calculated. The phonon modes responsible for the phase transition (upon heating) from high pressure phases to ambient pressure phases are identified. Moreover, ab initio lattice dynamics calculations in the framework of quasi-harmonic approximations are used to study the anisotropic thermal expansion behavior of γ-LiAlO2.
 Please wait while we load your content...
Please wait while we load your content...

