
Initial stage of atomic layer deposition of 2D-MoS2 on a SiO2 surface: a DFT study†

Abstract
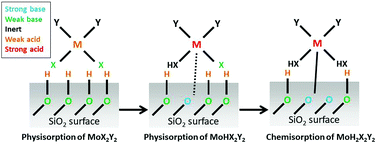
In this study, we investigate the reactions involving Atomic Layer Deposition (ALD) of 2D-MoS2 from the heteroleptic precursor Mo(NMe2)2(NtBu)2 and H2S as the co-reagent on a SiO2(0001) surface by means of density functional theory (DFT). All dominant reaction pathways from the early stage of adsorption of each ALD reagent to the formation of bulk-like Mo and S at the surface are identified. In the metal pulse, proton transfer from terminal OH groups on the SiO2 to the physisorbed metal precursor increases the Lewis acidity of Mo and Lewis basicity of O, which gives rise to the chemical adsorption of the metal precursor. Proton transfer from the surface to the dimethylamido ligands leads to the formation and desorption of dimethylamine. In contrast, the formation and desorption of tert-butylamine is not energetically favorable. The tert-butylimido ligand can only be partially protonated in the metal pulse. In the sulphur pulse, co-adsorption and dissociation of H2S molecules give rise to the formation and desorption of tert-butylamine. Through the calculated activation energies, the cooperation between H2S molecules (‘cooperative’ mechanism) is shown to have a profound influence on the formation and desorption of tert-butylamine, which are crucial steps in the initial ALD deposition of 2D-MoS2 on SiO2. The cyclic ALD reactions give rise to the formation of a buffer layer which might have important consequences for the electrical and optical properties on the 2D layer formed in the subsequent homodeposition.
 Please wait while we load your content...
Please wait while we load your content...

