
Ab initio calculation of energy levels of trivalent lanthanide ions†

Abstract
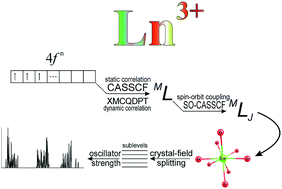
The energy levels of Ln3+ ions are known to be only slightly dependent on the ion environment. This allows one to predict the spectra of f–f transitions in Ln3+ complexes using group theory and simple semiempirical models: Russell–Saunders scheme for spin–orbit coupling, ligand-field theory for the splitting of the electronic levels, and Judd–Ofelt parameterization for reproducing the intensity of f–f transitions. Nevertheless, a fully ab initio computational scheme employing no empirical parameterization and suitable for any asymmetrical environment of Ln3+ would be instructive. Here we present such a scheme based on the multireference SA-CASSCF/XMCQPDT2/SO-CASSCF (state-averaged complete active space SCF, quasi-degenerate perturbation theory, and spin–orbit CASSCF) approach for trivalent lanthanide ions from Ce3+ (4f1) to Yb3+ (4f13). To achieve the most accurate results, we analyse the factors that influence the accuracy of the calculation: basis set size, state averaging scheme, effect of the low-spin states on the energy gap between the high-spin states (e.g., effect of triplets on the septet–quintet gaps in f6 or f8 configurations), and radial and angular correlations in the 4f shell. Our calculated energy levels agree well with the experimental values. We have shown that low-lying highest-spin and second-highest spin states are reproduced very well, while for higher-lying states the accuracy of the calculation decreases. The procedure was verified by calculating optical emission spectra of NaYF4:Eu,Tb; YAG:Eu,Tb; and Tb(acac)3bpm (bpm is 2,2′-bipyridine, acac is acetylacetonate, and YAG is yttrium aluminium garnet). For these compounds ligand-field induced electric-dipole transition intensities were calculated.
 Please wait while we load your content...
Please wait while we load your content...

