
Modeling the adsorption equilibrium of small-molecule gases on graphene: effect of the volume to surface ratio
 †a
and
Marco
Cecchini
*ab
†a
and
Marco
Cecchini
*ab
Abstract
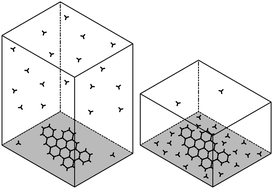
In most technological applications involving liquids or gases interacting with solids, the first event is the adsorption of molecules onto a solid surface. Here, we focus on the theoretical understanding of adsorption equilibrium at the solid–gas interface. In the limit of physisorption, we find that adsorption probability is independent of the initial concentration of monomers, whereas it varies with the available volume to surface ratio, which depends on the experimental setup. This theoretical finding is verified numerically by molecular dynamics simulations of five small-molecule gases physisorbing on graphene. The simulations provide quantitative estimates of the adsorption free energy, which are used to benchmark analytical and numerical integrations of the corresponding partition functions. The significance of the above theoretical result is analyzed in the context of molecular self-assembly at surfaces and interfaces. Our interpretation indicates that there exist (at least) two distinct pathways for 2D self-assembly, which may or may not involve the formation of a 2D disordered intermediate. Also, it predicts that the critical concentration for self-assembly may be shifted by varying the aspect ratio of the experimental setup.
 Please wait while we load your content...
Please wait while we load your content...

