
Energetics and dynamics of the non-natural fluorescent 4AP:DAP base pair†

Abstract
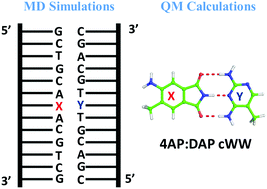
The fluorescent non-natural 4-aminophthalimide (4AP) base, when paired to the complementary 2,4-diaminopyrimidine (DAP) nucleobase, is accommodated in a B-DNA duplex being efficiently recognized and incorporated by DNA polymerases. To complement the experimental studies and rationalize the impact of the above non-natural bases on the structure, stability and dynamics of nucleic acid structures, we performed quantum mechanics (QM) calculations along with classical molecular dynamics (MD) simulations. QM calculations were initially focused on the geometry and energetics of the 4AP:DAP non-natural pair and of H-bonded base pairs between 4AP and all the natural bases in their classical Watson–Crick geometries. The QM calculations indicate that the 4AP:DAP pair, despite the fact that it can form 3 H-bonds in a classic Watson–Crick geometry, has a stability comparable to the A:T pair. Then, we extended the study to reverse Watson–Crick geometries, characteristic of parallel strands. MD simulations were carried out on two 13-mer DNA duplexes, featuring a central 4AP:DAP or A:T pair, respectively. No major structural deformation of the duplex was observed during the MD simulation. Snapshots from the MD simulations were subjected to QM calculations to investigate the 4AP:DAP interaction energy when embedded into a duplex structure, and to investigate the impact of the two non-natural bases on the stacking interactions with adjacent bases in the DNA duplex. We found a slight increase in stacking interactions involving the 4AP:DAP pair, counterbalanced by a moderate decrease in H-bonding interactions of the 4AP:DAP and of the adjacent base pairs in the duplex. The results of our study are in agreement with experimental data and complement them by providing an insight into which factors contribute positively and which factors contribute negatively to the structural compatibility of the fluorescent 4AP:DAP pair with a B-DNA structure.
 Please wait while we load your content...
Please wait while we load your content...

