
A computational study of CO oxidation reactions on metal impurities in graphene divacancies†
 ‡*a
Dongwei
Ma
*b
and
‡*a
Dongwei
Ma
*b
and
Abstract
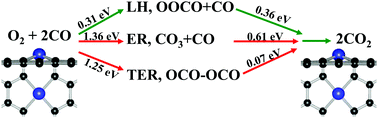
Based on the density functional theory calculations, the formation geometry, electronic properties, and catalytic activity of metal impurities in divacancy graphene (M-DG, M = Mo, Fe, Co, and Ni) were systematically investigated. It has been found that the reactive gases have different stabilities on M-DG substrates, and these quite stable substrates exhibit high catalytic activity for CO oxidation by comparing the traditional Eley–Rideal (ER) and Langmuir–Hinshelwood (LH), as well as the new termolecular ER (TER) mechanisms. For the Co-DG substrate, the coadsorption of O2 and CO as a starting step is an energetically more favorable process, whereas the dissociation reaction of O2 molecules on Mo-DG substrate has a much smaller energy barrier, and the generation of atomic oxygen is active for CO oxidation. These results indicate that the varied adsorption behaviors of reactive gases on M-DG substrates can determine the catalytic pathways and energy barriers, which give us insight into the surface reactivity of graphene–metal composite catalysis in energy-related devices.
 Please wait while we load your content...
Please wait while we load your content...

