
A new potential energy surface of the OH2+ system and state-to-state quantum dynamics studies of the O+ + H2 reaction†
 *ab
*ab
Abstract
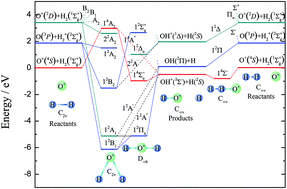
A new global potential energy surface (PES) of the O+ + H2 system was constructed with the permutation invariant polynomial neural network method, using about 63 000 ab initio points, which were calculated by employing the multi-reference configuration interaction method with aug-cc-pVTZ and aug-cc-pVQZ basis sets. For improving the accuracy of the PES, the basis set was extrapolated to the complete basis set limit by the two-point extrapolation method. The root mean square error of fitting was only 5.28 × 10−3 eV. The spectroscopic constants of the diatomic molecules were calculated and compared with previous theoretical and experimental results, which suggests that the present results agree well with the experiment. On the newly constructed PES, reaction dynamics studies were performed using the time-dependent wave packet method. The calculated integral cross sections (ICSs) were compared with the available theoretical and experimental results, where a good agreement with the experimental data was seen. Significant forward and backward scatterings were observed in the whole collision energy region studied. At the same time, the differential cross sections biased the forward scattering, especially at higher collision energies.
 Please wait while we load your content...
Please wait while we load your content...

