
First principles Monte Carlo simulations of unary and binary adsorption: CO2, N2, and H2O in Mg-MOF-74†
 a
Mansi S.
Shah,
ab
Jeffrey R.
Long,
cd
Michael
Tsapatsis
b
and
J. Ilja
Siepmann
*ab
a
Mansi S.
Shah,
ab
Jeffrey R.
Long,
cd
Michael
Tsapatsis
b
and
J. Ilja
Siepmann
*ab
Abstract
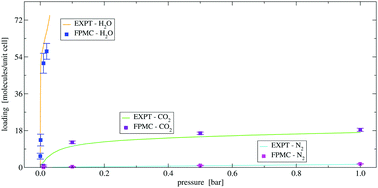
Dative bonding of adsorbate molecules onto coordinatively-unsaturated metal sites in metal–organic frameworks can lead to unique adsorption selectivities. However, the distortion of the electron density during dative bonding poses a challenge for force-field-based simulations. Here, we report first principles Monte Carlo simulations with the PBE-D3 functional for the adsorption of CO2, N2, and H2O in Mg-MOF-74, and obtain accurate predictions of the unary isotherms without any of the adjustments or fitting often required for systems with strong adsorption sites. Simulations of binary CO2/N2 and H2O/CO2 mixtures yield selectivities of 200 and 160, respectively, and indicate that predictions from ideal adsorbed solution theory need to be viewed with caution.
 Please wait while we load your content...
Please wait while we load your content...

