
Creep-induced anisotropy in covalent adaptable network polymers
 *a
*a
Abstract
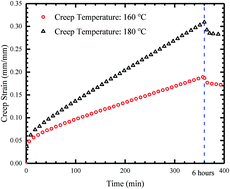
Anisotropic polymers with aligned macromolecule chains exhibit directional strengthening of mechanical and physical properties. However, manipulating the orientation of polymer chains in a fully cured thermoset is almost impossible due to its permanently crosslinked nature. In this paper, we demonstrate that rearrangeable networks with bond exchange reactions (BERs) can be utilized to tailor the anisotropic mechanical properties of thermosetting polymers. When a constant force is maintained at BER activated temperatures, the malleable thermoset creeps in the direction of stress, and macromolecule chains align themselves in the same direction. The aligned polymer chains result in an anisotropic network with a stiffer mechanical behavior in the direction of creep, while with a more compliant behavior in the transverse direction. The degree of network anisotropy is proportional to the amount of creep strain. A multi-length scale constitutive model is developed to study the creep-induced anisotropy of thermosetting polymers. The model connects the micro-scale BER kinetics, orientation of polymer chains, and directional mechanical properties of network polymers. Without any fitting parameters, it is able to predict the evolution of creep strain at different temperatures and anisotropic stress–strain behaviors of CANs after creep. Predictions on the chain orientation are verified by molecular dynamics (MD) simulation. Based on parametric studies, it is shown that the influences of creep time and temperature on the network anisotropy can be generalized into a single parameter, and the evolution of directional modulus follows an Arrhenius type time–temperature superposition principle (TTSP). The presented work provides a facile approach to transform isotropic thermosets into anisotropic ones using simple heating, and their directional properties can be readily tailored by the processing conditions.
 Please wait while we load your content...
Please wait while we load your content...

