
Machine learning for autonomous crystal structure identification
 a
Michael P.
Howard,
a
Andrew L.
Ferguson
bc
and
Athanassios Z.
Panagiotopoulos
*a
a
Michael P.
Howard,
a
Andrew L.
Ferguson
bc
and
Athanassios Z.
Panagiotopoulos
*a
Abstract
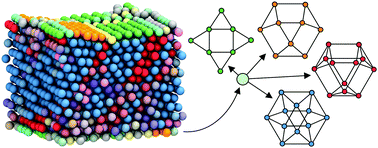
We present a machine learning technique to discover and distinguish relevant ordered structures from molecular simulation snapshots or particle tracking data. Unlike other popular methods for structural identification, our technique requires no a priori description of the target structures. Instead, we use nonlinear manifold learning to infer structural relationships between particles according to the topology of their local environment. This graph-based approach yields unbiased structural information which allows us to quantify the crystalline character of particles near defects, grain boundaries, and interfaces. We demonstrate the method by classifying particles in a simulation of colloidal crystallization, and show that our method identifies structural features that are missed by standard techniques.
 Please wait while we load your content...
Please wait while we load your content...

