
A framework for multi-scale simulation of crystal growth in the presence of polymers†
 a
William W.
Porter, III
c
and
a
William W.
Porter, III
c
and
Abstract
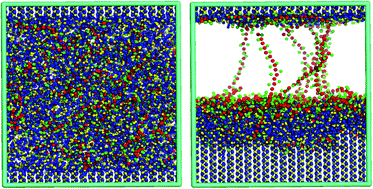
We present a multi-scale simulation method for modeling crystal growth in the presence of polymer excipients. The method includes a coarse-grained (CG) model for small molecules of known crystal structure whose force field is obtained using structural properties from atomistic simulations. This CG model is capable of stabilizing the molecular crystal structure and capturing the crystal growth from the melt for a wide range of small organic molecules, as demonstrated by application of our method to the molecules isoniazid, urea, sulfamethoxazole, prilocaine, oxcarbazepine, and phenytoin. This CG model can also be used to study the effect of additives, such as polymers, on the inhibition of crystal growth by polymers, as exemplified by our simulation of suppression of the rate of crystal growth of phenytoin, an active pharmaceutical ingredient (API), by a cellulose excipient, functionalized with acetate (Ac), hydroxy-propyl (Hp) and succinate (Su) groups. We show that the efficacy of the cellulosic polymers in slowing crystal growth of small molecules strongly depends on the functional group substitution on the cellulose backbone, with the acetate substituent group slowing crystal growth more than does the deprotonated succinate group, which we confirm by experimental drug supersaturation studies.
 Please wait while we load your content...
Please wait while we load your content...

