
Real single ion solvation free energies with quantum mechanical simulation†

Abstract
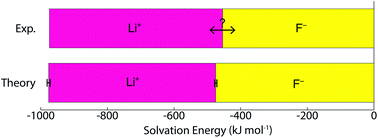
Single ion solvation free energies are one of the most important properties of electrolyte solutions and yet there is ongoing debate about what these values are. Only the values for neutral ion pairs are known. Here, we use DFT interaction potentials with molecular dynamics simulation (DFT-MD) combined with a modified version of the quasi-chemical theory (QCT) to calculate these energies for the lithium and fluoride ions. A method to correct for the error in the DFT functional is developed and very good agreement with the experimental value for the lithium fluoride pair is obtained. Moreover, this method partitions the energies into physically intuitive terms such as surface potential, cavity and charging energies which are amenable to descriptions with reduced models. Our research suggests that lithium's solvation free energy is dominated by the free energetics of a charged hard sphere, whereas fluoride exhibits significant quantum mechanical behavior that cannot be simply described with a reduced model.
 Please wait while we load your content...
Please wait while we load your content...

