
Dynamics of structural diffusion in phosphoric acid hydrogen-bond clusters†

Abstract
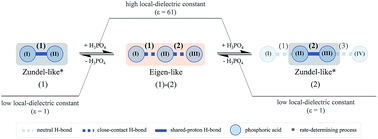
The dynamics and mechanism of structural diffusion in H3PO4 hydrogen-bond (H-bond) clusters were studied under excess proton conditions using ab initio calculations at the RIMP2/TZVP level and Born–Oppenheimer molecular dynamics (BOMD) simulations. The outstanding feature of our theoretical approach was the use of combined vibrational and NMR spectroscopic techniques that enable local dynamical motions and their activation energies to be studied in detail. Based on the concept of presolvation, H+(H3PO4)n (n = 2–5) clusters were selected as model systems, in which the rate-determining intermediate complexes in the structural diffusion process were studied in detail. The potential energy curves for proton displacement suggested that Zundel-like complexes are preferentially formed in linear H-bond chains in low local-dielectric environments, whereas Eigen-like complexes are stabilized in high local-dielectric environments. The RIMP2/TZVP results revealed that the Eigen–Zundel–Eigen mechanism, which leads to a positive charge displacement, is a short-range process (n = 2–3), and structural diffusion is enhanced by fluctuations in the H-bond chain length and local-dielectric environment. The vibrational and 1H NMR spectra obtained from BOMD simulations confirmed the proposed mechanism and suggested that for protonated H3PO4 clusters, structural diffusion can proceed without the reorientation of H3PO4 molecules (frustrated rotation), as in the case of neat liquid H3PO4. Because the H+ species cannot be directly probed in experiments, this theoretical study suggested for the first time characteristic properties of exchanging protons in H3PO4 systems under excess proton conditions, which can be used to assist experimental investigations.
 Please wait while we load your content...
Please wait while we load your content...

