
Machine learnt bond order potential to model metal–organic (Co–C) heterostructures†
 ‡*a
Fatih G.
Sen,
a
Maria K. Y.
Chan
a
and
Subramanian K. R. S.
Sankaranarayanan
*a
‡*a
Fatih G.
Sen,
a
Maria K. Y.
Chan
a
and
Subramanian K. R. S.
Sankaranarayanan
*a
Abstract
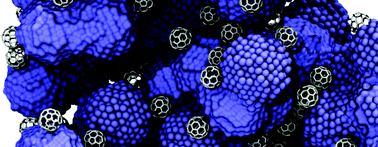
A fundamental understanding of the inter-relationships between structure, morphology, atomic scale dynamics, chemistry, and physical properties of mixed metallic-covalent systems is essential to design novel functional materials for applications in flexible nano-electronics, energy storage and catalysis. To achieve such knowledge, it is imperative to develop robust and computationally efficient atomistic models that describe atomic interactions accurately within a single framework. Here, we present a unified Tersoff–Brenner type bond order potential (BOP) for a Co–C system, trained against lattice parameters, cohesive energies, equation of state, and elastic constants of different crystalline phases of cobalt as well as orthorhombic Co2C derived from density functional theory (DFT) calculations. The independent BOP parameters are determined using a combination of supervised machine learning (genetic algorithms) and local minimization via the simplex method. Our newly developed BOP accurately describes the structural, thermodynamic, mechanical, and surface properties of both the elemental components as well as the carbide phases, in excellent accordance with DFT calculations and experiments. Using our machine-learnt BOP potential, we performed large-scale molecular dynamics simulations to investigate the effect of metal/carbon concentration on the structure and mechanical properties of porous architectures obtained via self-assembly of cobalt nanoparticles and fullerene molecules. Such porous structures have implications in flexible electronics, where materials with high electrical conductivity and low elastic stiffness are desired. Using unsupervised machine learning (clustering), we identify the pore structure, pore-distribution, and metallic conduction pathways in self-assembled structures at different C/Co ratios. We find that as the C/Co ratio increases, the connectivity between the Co nanoparticles becomes limited, likely resulting in low electrical conductivity; on the other hand, such C-rich hybrid structures are highly flexible (i.e., low stiffness). The BOP model developed in this work is a valuable tool to investigate atomic scale processes, structure–property relationships, and temperature/pressure response of Co–C systems, as well as design organic–inorganic hybrid structures with a desired set of properties.
 Please wait while we load your content...
Please wait while we load your content...

