
Relativistic DFT investigation of electronic structure effects arising from doping the Au25 nanocluster with transition metals†
 bc
and
Christine M.
Aikens
*a
bc
and
Christine M.
Aikens
*a
Abstract
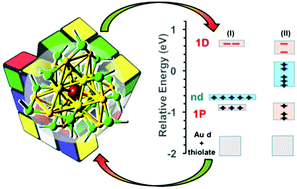
We perform a theoretical investigation using density functional theory (DFT) and time-dependent DFT (TDDFT) on the doping of the Au25(SR)18−1 nanocluster with group IX transition metals (M = cobalt, rhodium and iridium). Different doping motifs, charge states and spin multiplicities were considered for the single-atom doped nanoclusters. Our results show that the interaction (or the lack of interaction) between the d-type energy levels that mainly originate from the dopant atom and the super-atomic levels plays an important role in the energetics, the electronic structure and the optical properties of the doped systems. The evaluated MAu24(SR)18q (q = −1, −3) systems favor an endohedral disposition of the doping atom typically in a singlet ground state, with either a 6- or 8-valence electron icosahedral core. For the sake of comparison, the role of the d energy levels in the electronic structure of a variety of doped Au25(SR)18−1 nanoclusters was investigated for dopant atoms from other families such as Cd, Ag and Pd. Finally, the effect of spin–orbit coupling (SOC) on the electronic structure and absorption spectra was determined. The information in this study regarding the relative energetics of the d-based and super-atom energy levels can be useful to extend our understanding of the preferred doping modes of different transition metals in protected gold nanoclusters.
- This article is part of the themed collection: 2017 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

