
High thermoelectric performances of monolayer SnSe allotropes†
 ‡a
Xiao-Hong
Shao
*a
‡a
Xiao-Hong
Shao
*a
Abstract
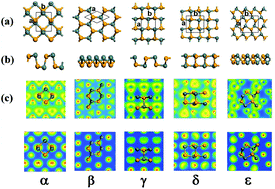
α-SnSe is one of the most promising thermoelectric materials with low thermal conductivity and a high power factor. Since the thermoelectric properties of a material have a strong dependence on its crystal structure, we study the energetic and thermoelectric properties of four new monolayer phases of SnSe (β, γ, δ and ε) together with α-SnSe using the ab initio density functional theory method. The calculated electronic structures show that all five phases are semiconductors with different band gaps. The α, β, γ, and δ phases have an indirect band gap with the hybridization of sp2 orbitals, whereas the ε phase has a direct band with the hybridization of sp3 orbitals. The thermoelectric transport properties and coefficients are obtained from the electronic structure using semi-classical Boltzmann theory, and the results indicate that the four new phases of SnSe (β, γ, δ and ε) all have better thermoelectric properties compared with the reported α phase. The predicted ZT value for the β-SnSe phase is 2.06 at 300 K, suggesting that it has great potential for novel thermoelectric applications.
 Please wait while we load your content...
Please wait while we load your content...

