
Computational study on C–B homolytic bond dissociation enthalpies of organoboron compounds†
 *a
*a
Abstract
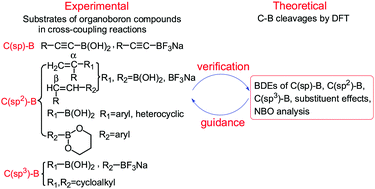
Based on many transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions of organoboron compounds in which C–B cleavages are involved, it is meaningful to understand one of the thermodynamic properties of the C–B bond, the strength of the C–B bond, which can be measured using homolytic bond dissociation enthalpies (BDEs). To this end, we first calculated 64 C–B BDEs of organoboron compounds by theoretical methods including composite high-level ab initio methods of G3, G4, G3B3, CBS-Q, CBS-QB3, and CBS-4M and 34 density functional theory (DFT) methods. The results show that it is reasonable and credible to regard the average values of six composite high-level methods as the standard C–B BDE values. By comparing the DFT methods, it is found that the M06-HF method provides the most accurate results and the root mean square error (RMSE) is the smallest of 6.4 kJ mol−1. Therefore, the C–B BDEs including C(sp)–B, C(sp2)–B and C(sp3)–B of organoboron compounds such as boronic acids, trifluoroborate salts, boronate esters, etc. as well as the substituent effects were investigated by using the M06-HF method. The results indicated that the different substituents including electron-donating groups (EDGs), electron-withdrawing groups (EWGs) and conjugated effect groups (CEGs) exhibit different effects on different types of C–B BDEs. Moreover, the natural bond orbital (NBO) analysis was performed in order to further disclose the essence of BDE change patterns.
 Please wait while we load your content...
Please wait while we load your content...

