
Studying physisorption processes and molecular friction of cycloparaphenylene molecules on graphene nano-sized flakes: role of π⋯π and CH⋯π interactions

Abstract
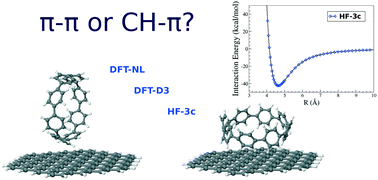
We theoretically study, by means of dispersion-corrected and cost-effective methods, the strength of non-covalent interactions between cyclic organic nanorings (i.e. [8]cycloparaphenylene molecule) and nano-sized (e.g. C96H24) graphene flakes acting as substrates. Both CH⋯π and π⋯π driven interactions are investigated, according to the relative orientation between the two weakly interacting monomers, whose potential energy profiles are accurately calculated in both cases. These configurations provide different physisorption curves, with the CH⋯π interaction leading to a larger well depth, and are found to slightly depend on edge effects of the nano-sized graphene flakes. Additionally, we fit the energy profiles to a compact (analytical) potential function, and study the atomic-scale friction between the molecule and the surface in the search of mechanisms for new molecular machines.
 Please wait while we load your content...
Please wait while we load your content...

