
Density functional theory studies on the conversion of hydroxyheme to iron-verdoheme in the presence of dioxygen†
 *a
and
*a
and
Abstract
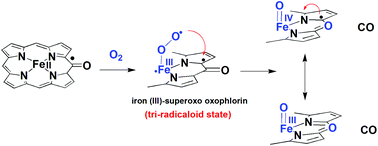
Detailed insight into the second step of heme degradation by heme oxygenase, oxophlorin to verdoheme and biliverdin, is presented. Density functional theory methods are reported for the conversion of oxophlorin to verdoheme. Since it is currently unclear whether dioxygen binding to iron oxophlorin is followed by a reduction or not, in this work we have focused on the difference in reactivity between [(Im)(O2˙)FeIII(PO˙)] (PO˙ is the oxophlorin dianion radical) and [(Im)(O2˙)FeIII(PO)]− (PO is the oxophlorin trianion). Thus, we have shown that in [(Im)(O2˙)FeIII(PO˙)] and [(Im)(O2˙)FeIII(PO)]−, the mechanisms are stepwise with an initial C–O bond activation to form a ring-structure where the oxophlorin is distorted from planarity. This is followed by homolytic dioxygen bond breaking that directly leads to iron–oxo verdoheme products. The [(Im)(O2˙)FeIII(PO˙)] mechanism proceeds via two-state-reactivity patterns on the adjacent doublet and quartet spin state surfaces, whereas the [(Im)(O2˙)FeIII(PO)]− route shows single-state-reactivity on a triplet spin state surface. In both, the rate determining step is the C–O bond activation, with substantially lower barriers on the [(Im)(O2˙)FeIII(PO˙)] surface of 12.15 kcal mol−1 in the gas phase compared to 22.55 kcal mol−1 for the intermediate-spin of [(Im)(O2˙)FeIII(PO)]−. The complete active space self-consistent-field wave functions with second-order multi-reference perturbation theory were also studied. Finally, the effects of the solvent and the medium on the reaction barriers were tested and shown to be considerable.
 Please wait while we load your content...
Please wait while we load your content...

