
Stanene based gas sensors: effect of spin–orbit coupling†
 *ab
*ab
Abstract
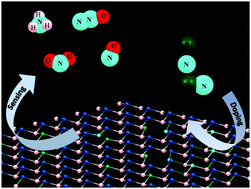
Density functional theory calculations are performed to investigate the gas sensing properties (NO, NO2, NH3 and N2O) of pure and doped (B@, N@, and B–N@) stanene. Dispersion corrected (DFT-D3) density functional calculations show that doping improves the interaction between stanene and gas molecules. The extent of interaction between the system and gas molecules is further studied through charge density difference (CDD), electrostatic potential (ESP) and Bader charge analysis. The electronic properties of pure stanene + gases are studied with and without the effect of spin–orbit coupling. Stanene + gas systems show the Rashba-type of spin-splitting under spin–orbit coupling (SOC), which is very promising for spintronic applications. Interestingly, the doped systems (B@-, N@-, and B–N@stanene) show higher selectivity and sensitivity toward gas molecules compared to pure stanene. Therefore, the B@-, N@-, and B–N@stanene systems are promising for semiconductor based gas sensors.
 Please wait while we load your content...
Please wait while we load your content...

