
DFT investigation of the interaction between single-walled carbon nanotubes and fluorene-based conjugated oligomers†

Abstract
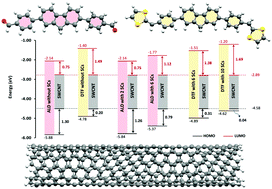
π-Conjugated oligomers with relatively short molecular backbones can be used effectively in dispersion of carbon nanotubes (CNTs). In this paper, we present a systematic study on interactions between diphenylene-fluorene oligomers (DPFs) and single-walled CNTs (SWCNTs) using density functional theory (DFT) calculations. Four DFT methods are used in this work: the long range (LR)-corrected CAM-B3LYP, the dispersion (D)-corrected B97D, the LR- and D-corrected wB97XD, and the hybrid B3LYP. The DPFs examined in this study contain different functional groups attached to the π-conjugated backbone, including two different end groups, carboxaldehyde (ALD) and dithiafulvenyl (DTF), and three different side chains (SCs), C8H17, OC10H21, and SC10H21. The computational results disclose the effects of end groups, SCs, and DFT methods on structures, dipole moments, and energetics of isolated DPFs and DPF/SWCNT combinations. Consistent with our previous study (involving oligo(p-phenylene ethynylene)s (OPEs)) [Aljohani et al., J. Phys. Chem. C, 2017, 121, 4692–4702], our results herein demonstrate that the type of end group plays a key role in determining the strength of interactions between SWNTs and conjugated oligomers. In particular, DTF-endcapped oligomers have a stronger electrostatic interaction with SWCNT than ALD-endcapped oligomers do. As a result, DTF-endcapped conjugated oligomers become more polarized than ALD-endcapped oligomers after complexing with SWCNTs. The magnitude of binding energy, on the other hand, shows dependence on the orientation of the backbone and side chains of these oligomers relative to the SWCNT which in the case of fluorene-based oligomers is not always favourable for optimal binding. This study indicates that fluorene-based oligomers might not be as good dispersants of SWCNTs as OPEs.
 Please wait while we load your content...
Please wait while we load your content...

