
Validation of density functionals for pancake-bonded π-dimers; dispersion is not enough†
 a
Miklos
Kertesz
*a
a
Miklos
Kertesz
*a
Abstract
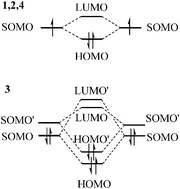
π-Stacking pancake bonding between radicals poses special challenges to density functional theories (DFTs) due to their shorter than van der Waals contact distances, their multireference singlet ground states and the concurrently important dispersion interactions. We examined over 50 DFTs including 22 with dispersion corrections on four different π-dimerized pancake-bonded systems exploring the performances of these DFTs in the very short intermolecular contact regime. We examined crucial energetic as well as geometric parameters against available high-level multireference average quadratic coupled cluster (MR-AQCC) results. Overall we did not find an omnipotent DFT applicable for all four pancake-bonded π-dimers. However, some DFTs were found to perform well for each individual system: M05-2X and PBE0-MBD are the only DFTs that work well for the phenalenyl π-dimer; BLYP is the only appropriate DFT for the 1,2,4,6-thiatriazine π-dimer; O3LYP works best on the double pancake-bonded 1,3,2,4,6-dithiatriazine π-dimer with a few acceptable ones and MN15L is the best method for K+TCNE− π-dimer in addition to a few acceptable ones. We believe our findings can deliver insights towards the design and characterization of the pancake-bond based materials and the development of new DFTs.
 Please wait while we load your content...
Please wait while we load your content...

