
Wheel-like, elongated, circular, and linear geometries in boron-based CnB7−n (n = 0–7) clusters: structural transitions and aromaticity†
 *a
*a
Abstract
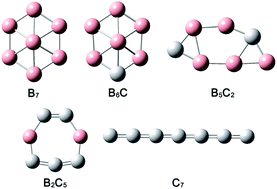
We report a quantum chemical study on the structural and bonding properties of a series of boron–carbon mixed clusters with seven atoms: CnB7−n (n = 0–7). Global-minimum structures were searched using the Coalescence Kick (CK) method, followed by B3LYP/6-311+G(d) calculations for full optimizations and energetics. Top candidate structures were further benchmarked at the single-point CCSD(T) level. Structural transitions were revealed to occur successively between wheel-like, elongated, circular, and linear geometries upon the increase of C contents in the clusters. Chemical bonding was elucidated via canonical molecular orbital (CMO) analyses and adaptive natural density partitioning (AdNDP). The number of delocalized electrons (σ plus π) in the clusters was shown to vary by one at a time from 5σ to 7σ, as well as from 3π to 6π, which allows aromaticity, antiaromaticity, and conflicting aromaticity to be precisely tuned according to the (4n + 2) and 4n Hückel rules. Delocalized π and σ bonds and their electron counting appear to dictate the cluster structures of the whole series. Aromaticity in the systems was independently confirmed using nucleus-independent chemical shifts (NICSs). The monocyclic B2C5 cluster was shown to possess the greatest NICS values, consistent with its 6π plus 6σ electron countings for double aromaticity. Our analyses also shed light on the reason why C in the filled-hexagonal B6C cluster occupies a peripheral site rather than the center and why C avoids hypercoordination in B–C binary clusters. A similar argument should be valid for other B–C clusters in prior reports, such as B6C2−, B7C−, and B8C.
 Please wait while we load your content...
Please wait while we load your content...

