
Hydrogen adsorption on MoS2-surfaces: a DFT study on preferential sites and the effect of sulfur and hydrogen coverage†

Abstract
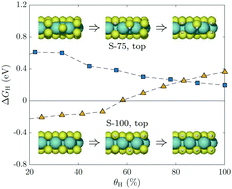
We report a comprehensive computational study of the intricate structure–property relationships governing the hydrogen adsorption trends on MoS2 edges with varying S- and H-coverages, as well as provide insights into the role of individual adsorption sites. Additionally, the effect of single- and dual S-vacancies in the basal plane on the adsorption energetics is assessed, likewise with an emphasis on the H-coverage dependency. The employed edge/site-selective approach reveals significant variations in the adsorption free energies, ranging between ∼±1.0 eV for the different edges-types and S-saturations, including differences of even as much as ∼1.2 eV between sites on the same edge. The incrementally increasing hydrogen coverage is seen to mainly weaken the adsorption, but intriguingly for certain configurations a stabilizing effect is also observed. The strengthened binding is seen to be coupled with significant surface restructuring, most notably the splitting of terminal S2-dimers. Our work links the energetics of hydrogen adsorption on 2H-MoS2 to both static and dynamic geometrical features and quantifies the observed trends as a function of H-coverage, thus illustrating the complex structure/activity relationships of the MoS2 catalyst. The results of this systematical study aims to serve as guidance for experimentalists by suggesting feasible edge/S-coverage combinations, the synthesis of which would potentially yield the most optimally performing HER-catalysts.
 Please wait while we load your content...
Please wait while we load your content...

