
Direct folding simulation of helical proteins using an effective polarizable bond force field†
 *bcd
*bcd
Abstract
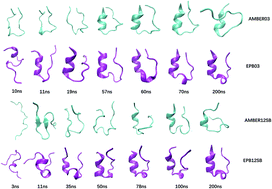
We report a direct folding study of seven helical proteins (2I9M, Trpcage, 1WN8, C34, N36, 2KES, 2KHK) ranging from 17 to 53 amino acids through standard molecular dynamics simulations using a recently developed polarizable force field-Effective Polarizable Bond (EPB) method. The backbone RMSDs, radius of gyrations, native contacts and native helix content are in good agreement with the experimental results. Cluster analysis has also verified that these folded structures with the highest population are in good agreement with their corresponding native structures for these proteins. In addition, the free energy landscape of seven proteins in the two dimensional space comprised of RMSD and radius of gyration proved that these folded structures are indeed of the lowest energy conformations. However, when the corresponding simulations were performed using the standard (nonpolarizable) AMBER force fields, no stable folded structures were observed for these proteins. Comparison of the simulation results based on a polarizable EPB force field and a nonpolarizable AMBER force field clearly demonstrates the importance of polarization in the folding of stable helical structures.
 Please wait while we load your content...
Please wait while we load your content...

