
Effects of the locality of a potential derived from hybrid density functionals on Kohn–Sham orbitals and excited states†
 *a
*a
Abstract
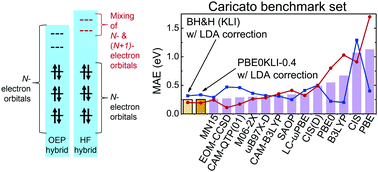
Density functional theory (DFT) has been an essential tool for electronic structure calculations in various fields. In particular, its hybrid method including the Hartree–Fock (HF) exchange term remarkably improves the reliability of DFT for chemical applications and computational material design. There are two different types of exchange–correlation potential that can be derived from hybrid functionals. The conventional approach adopts a non-multiplicative potential including the non-local HF exchange operator. Herein, we propose to use a local multiplicative potential as an alternative for accurate excited state calculations. We show that such a local potential can be derived from existing global hybrid functionals using the optimized effective potential method. As a proof-of-concept, we chose PBE0 and investigated its performance for the Caricato benchmark set. Unlike the conventional one, the local potential produced orbital energy gaps with no strong dependence on the mixing ratio as a good approximation for optical excitations. Furthermore, its time-dependent DFT resulted in a surprisingly small mean absolute error even with a local density approximation kernel, surpassing all reported values with various popular functionals. In particular, most excitations were dictated by single orbital transitions due to physically meaningful virtual orbitals, which is beneficial to clear interpretations in the molecular orbital picture.
 Please wait while we load your content...
Please wait while we load your content...

