
Photoinduced bimolecular electron transfer from aromatic amines to pentafluorophenyl porphyrin combined with ultrafast charge recombination persistence with Marcus inverted region†

Abstract
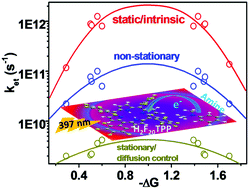
The dynamics of photoinduced bimolecular reductive electron transfer between meso-tetrakis(pentafluorophenyl)porphyrin (H2F20TPP), an acceptor (A), and five aromatic amines (donor (D)) with varying oxidation potentials (aniline (AN), N-methylaniline (MAN), N-ethylaniline (EAN), N,N-dimethylaniline (DMAN) and N,N-diethylaniline (DEAN)) in dichloromethane (DCM) as a solvent as well as in neat donor solvents were investigated by employing nanosecond to femtosecond time-resolved fluorescence spectroscopy and femtosecond time-resolved transient absorption spectroscopy upon S2 excitation of H2F20TPP. Systematic studies of time-resolved fluorescence quenching dependent on the donor concentration in the concentration range of 0.01–2 M and finally in neat donor solvents broadly enabled us to determine the electron transfer dynamics in three regimes of electron transfer: stationary or diffusion-controlled electron transfer, non-stationary electron transfer and intrinsic or ultrafast electron transfer. Depending upon the electron-donating ability of the studied donors, intrinsic electron transfer was found to occur in the time domain of ∼1–9 ps and diffusion-controlled ET dynamics was observed in the time domain of 200–500 ps, whereas the maximum limit of non-stationary electron transfer could be observed in the time domain of 15–50 ps. Femtosecond transient absorption studies together with global and target analysis helped to identify the spectral signature of the (H2F20TPP˙−) radical anion as the product of ET. To the best of our knowledge, this is the first ever evidence that shows the spectra of an anion as the product of ET for any porphyrin-based electron transfer dynamics. However, transient absorption measurements confirm that intrinsic ET occurs in the Qy state, whereas diffusion-controlled ET occurs in the hot Qx as well as in the thermal equilibrium Qx state. The most remarkable fact derived from the measurements of transient absorption was that the rate of the forward electron transfer (CS) is exactly the same as the rate of the backward electron transfer (CR) for all three regimes of ET. The thermodynamic driving force for CR was found to lie in the range of the total reorganization energy for the studied systems and hence falls in the Marcus optimal region, and the CR process is barrierless. The dependence on the driving force of the combination of forward and reverse electron transfer exhibited a bell-shaped curve for all three regimes of electron transfer, even though the rate of intrinsic ET is higher by a factor of ∼102 than that of diffusion-controlled ET. These results unambiguously favour the Marcus theory, in particular the controversial Marcus inverted region, of outer-sphere electron transfer.
 Please wait while we load your content...
Please wait while we load your content...

