
Towards open boundary molecular dynamics simulation of ionic liquids
 a
and
a
and
Abstract
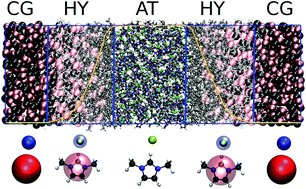
We extend the use of the adaptive resolution (AdResS) method in its grand canonical-like version (GC-AdResS) to the molecular dynamics simulation of 1,3-dimethylimidazolium chloride. We show that the partitioning of the total system in a subsystem of interest with atomistic details and a reservoir of coarse-grained particles leads to satisfactory results. The challenging aspect of this study, compared to previous AdResS simulations, is the presence of charged particles and the necessity of addressing the question about the minimal physical input needed to model the coarse-grained particles in the reservoir. We propose two different approaches and show that in both cases they are sufficient to capture the decisive physical characteristics that allow a valid system–reservoir coupling. The technically satisfactory results pave the way for the multiscale analysis of ionic liquids and truly open boundary molecular simulations.
 Please wait while we load your content...
Please wait while we load your content...

