
Computational study of the interplay between intermolecular interactions and CO2 orientations in type I hydrates
Abstract
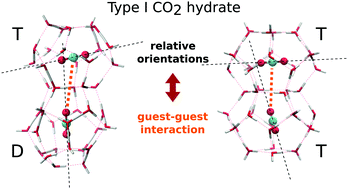
Carbon dioxide (CO2) molecules show a rich orientation landscape when they are enclathrated in type I hydrates. Previous studies have described experimentally their preferential orientations, and some theoretical works have explained, but only partially, these experimental results. In the present paper, we use classical molecular dynamics and electronic density functional theory to advance in the theoretical description of CO2 orientations within type I hydrates. Our results are fully compatible with those previously reported, both theoretical and experimental, the geometric shape of the cavities in hydrate being, and therefore, the steric constraints, responsible for some (but not all) preferential angles. In addition, our calculations also show that guest–guest interactions in neighbouring cages are a key factor to explain the remaining experimental angles. Besides the implication concerning equation of state hydrate modeling approximations, the conclusion is that these guest–guest interactions should not be neglected, contrary to the usual practice.
 Please wait while we load your content...
Please wait while we load your content...

