
Probing the electronic structures of Con (n = 1–5) clusters on γ-Al2O3 surfaces using first-principles calculations†
 *ab
*ab
Abstract
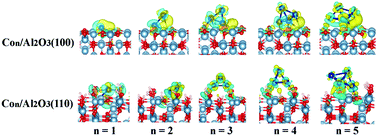
The electronic structure of the oxide-supported nanoclusters based on metal–support interactions including charge and spin reorganization plays a crucial role in the catalytic activity at the interface region. In this work, by using density functional theory periodic calculations, we theoretically investigated the stability, nucleation and electronic properties of cobalt clusters Con (n = 1–5) supported on γ-Al2O3 surfaces including dehydrated (100) and hydrated (110) surfaces. In Con/γ-Al2O3(100) (n = 1–5) and Con/γ-Al2O3(110) (n = 1 and 2), the Con clusters prefer to adsorb on the surface. However, for Con/γ-Al2O3(110) (n = 3–5), the Con clusters bind to surface hydroxyl groups directly. Our results revealed that due to the metal–support interaction, small Co clusters strongly adsorb onto the surfaces. Nucleation energy disclosed that the critical cluster sizes are 4 and 3 for the dehydrated (100) and hydrated (110) surfaces, respectively, suggesting that the presence of γ-Al2O3 surfaces remarkably affects and even prevents the sintering process of small Co clusters. More importantly, the charge analysis showed that both metal cluster and support play important roles in the charge state of clusters.
 Please wait while we load your content...
Please wait while we load your content...

