
Flexibility in metal–organic frameworks derived from positional and electronic effects of functional groups†

Abstract
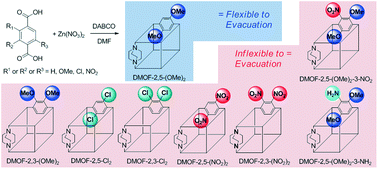
The position of identical functional groups and the subsequent electron density of structural benzene rings in a zinc-based metal–organic framework (MOF) have been controlled to reveal flexibility (or breathing behavior) differences. Both ortho- and para-positioned bi-functional benzene-1,4-dicarboxylic acid (BDC) ligands were synthesized with amino-, chloro-, methoxy-, and nitro groups. Additionally, two tri-functionalized, dimethoxy-amino and dimethoxy-nitro BDCs were prepared. All bi- and tri-functionalized BDCs were successfully incorporated into DABCO MOFs (DMOFs), except two diamino BDCs which were insoluble and thermally unstable. Among the eight bi-/tri-functionalized DMOFs, only para-dimethoxy exhibited flexibility in its framework after evacuation in preparation for N2 isotherm measurement. Since the dimethoxy combination has the most electron-rich environment in the benzene ring of the BDC in this series, this indicates that electron density plays a role in the flexibility changes of identically bi-functionalized DMOFs. However, the electron density alone could not fully explain the flexibility changes suggesting that the position of the functional groups is also important. These findings have been corroborated through the synthesis of two tri-functionalized DMOFs with identical functional group locations but opposite electronic environments.
 Please wait while we load your content...
Please wait while we load your content...

