
Ion mobility-resolved collision-induced dissociation and electron transfer dissociation of N-glycopeptides: gathering orthogonal connectivity information from a single mass-selected precursor ion population

Abstract
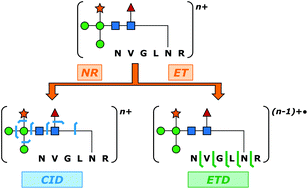
Glycopeptide-level mass spectrometry (MS) and tandem mass spectrometry (MS/MS) analyses are commonly performed to establish site-specific protein glycosylation profiles that are of central importance to gaining structure–function insights on glycoproteins. Confoundingly, the complete characterization of glycopeptide connectivity usually requires the acquisition of multiple MS/MS fragmentation spectra. Complementary ion fragmentation techniques such as collision-induced dissociation (CID) and electron transfer dissociation (ETD) are often applied in concert to address this need. While structurally informative, the requirement for acquisition of two MS/MS spectra per analyte places considerable limitations upon the breadth and depth of large-scale glycoproteomic inquiry. Here, a previously developed method of multiplexing CID and ETD is applied to the study of glycopeptides for the first time. Integration of the two dissociation methods was accomplished through addition of an ion mobility (IM) dimension that disperses the two stages of MS/MS in time. This allows the two MS/MS spectra to be acquired within a few milliseconds of one another, and to be deconvoluted in post-processing. Furthermore, the method allows both fragmentation readouts to be obtained from the same precursor ion packet, thus reducing the inefficiencies imposed by separate CID and ETD acquisitions and the relatively poor precursor ion to fragment ion conversion typical of ETD. N-Linked glycopeptide ions ranging in molecular weight from 1.8 to 6.5 kDa were generated from four model glycoproteins that collectively encompassed paucimannosidic, high mannose, and complex types of N-glycosylation. In each case, IM-resolved CID and ETD events provided complete coverage of the glycan topology and peptide sequence coverages ranging from 48.4% (over 32 amino acid residues) to 85.7% (over eight amino acid residues). The potential of this method for large-scale glycoproteomic analysis is discussed.
 Please wait while we load your content...
Please wait while we load your content...

