
Ultrafast dynamics of formation and autodetachment of a dipole-bound state in an open-shell π-stacked dimer anion†

Abstract
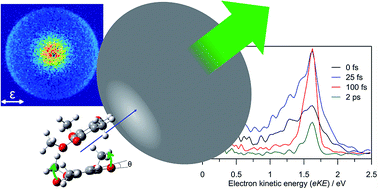
Isolated π-stacked dimer radical anions present the simplest model of an excess electron in a π-stacked environment. Here, frequency-, angle-, and time-resolved photoelectron imaging together with electronic structure calculations have been used to characterise the π-stacked coenzyme Q0 dimer radical anion and its exited state dynamics. In the ground electronic state, the excess electron is localised on one monomer with a planar para-quinone ring, which is solvated by the second monomer in which carbonyl groups are bent out of the para-quinone ring plane. Through the π-stacking interaction, the dimer anion exhibits a number of charge-transfer (intermolecular) valence-localised resonances situated in the detachment continuum that undergo efficient internal conversion to a cluster dipole-bound state (DBS) on a ∼60 fs timescale. In turn, the DBS undergoes vibration-mediated autodetachment on a 2.0 ± 0.2 ps timescale. Experimental vibrational structure and supporting calculations assign the intermolecular dynamics to be facilitated by vibrational wagging modes of the carbonyl groups on the non-planar monomer. At photon energies ∼0.6–1.0 eV above the detachment threshold, a competition between photoexcitation of an intermolecular resonance leading to the DBS, and photoexcitation of an intramolecular resonance leading to monomer-like dynamics further illustrates the π-stacking specific dynamics. Overall, this study provides the first direct observation of both internal conversion of resonances into a DBS, and characterisation of a vibration-mediated autodetachment in real-time.
 Please wait while we load your content...
Please wait while we load your content...

