
Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(iii) ylides†

Abstract
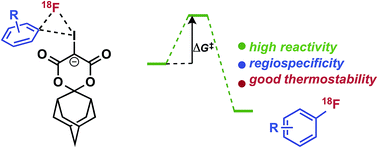
Synthesis of non-activated electron-rich and sterically hindered 18F-arenes remains a major challenge due to limitations of existing radiofluorination methodologies. Herein, we report on our mechanistic investigations of spirocyclic iodonium(III) ylide precursors for arene radiofluorination, including their reactivity, selectivity, and stability with no-carrier-added [18F]fluoride. Benchmark calculations at the G2[ECP] level indicate that pseudorotation and reductive elimination at iodine(III) can be modeled well by appropriately selected dispersion-corrected density functional methods. Modeling of the reaction pathways show that fluoride–iodonium(III) adduct intermediates are strongly activated and highly regioselective for reductive elimination of the desired [18F]fluoroarenes (difference in barriers, ΔΔG‡ > 25 kcal mol−1). The advantage of spirocyclic auxiliaries is further supported by NMR spectroscopy studies, which bolster evidence for underlying decomposition processes which can be overcome for radiofluorination of iodonium(III) precursors. Using a novel adamantyl auxiliary, sterically hindered iodonium ylides have been developed to enable highly efficient radiofluorination of electron-rich arenes, including fragments of pharmaceutically relevant nitrogen-containing heterocycles and tertiary amines. Furthermore, this methodology has been applied for the syntheses of the radiopharmaceuticals 6-[18F]fluoro-meta-tyrosine ([18F]FMT, 11 ± 1% isolated radiochemical yield, non-decay-corrected (RCY, n.d.c.), n = 3), and meta-[18F]fluorobenzylguanidine ([18F]mFBG, 14 ± 1% isolated RCY, n.d.c., n = 3) which cannot be directly radiolabeled using conventional nucleophilic aromatic substitution with [18F]fluoride.
 Please wait while we load your content...
Please wait while we load your content...

