
Molecular dynamics simulation elucidates the preferential binding affinity of sodium and tetramethylammonium ions for tetrameric Nafion unit under aqueous conditions†
Abstract
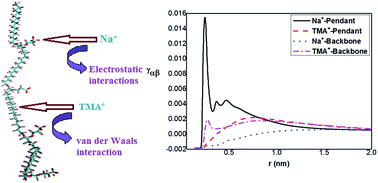
Perfluorinated Nafions are highly conductive to positively charged cations, making themselves useful for suitable membrane applications. In this paper, using atomistic molecular dynamics simulations we show that smaller sodium ions (Na+) prefer to stay in the vicinity of the hydrophilic pendant groups of the Nafion owing to their strong electrostatic interactions with the negatively charged sulfonate groups of Nafion. On the contrary, bulkier tetramethylammonium ions (TMA+) mostly prefer to accommodate themselves around the hydrophobic backbone of the membrane due to strong hydrophobic van der Waals interactions between its methyl groups and the –CF2 linkages of the backbone. We also investigate the thermodynamic driving forces facilitating such preferential binding interactions and it turns out that majority of these interactions are entropically favourable. The preferential uptake of a given ionic species by Nafion ionomer has been shown to further affect the translational dynamics of the water molecules present in its first solvation shell.
 Please wait while we load your content...
Please wait while we load your content...

