
Microhydration effects on the structures and electrophilic properties of cytidine†
Abstract
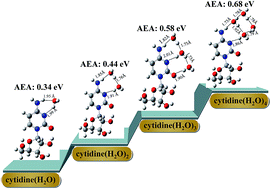
Microhydration effects on the geometrical structures, electron affinities and charge distributions of cytidine and their anions have been investigated systematically using density functional theory (DFT), by explicitly considering cytidine complexes with up to four water molecules. Various structures of neutral and anionic cytidine(H2O)n (n = 2–4) have been predicted, and N3, H–N4 and O2 are found to be the most favorable water-binding sites of cytidine. The adiabatic electron affinities of cytidine(H2O)n increase linearly with the number of hydrating water molecules, indicating that they would obtain a stronger ability to attract electrons with the hydration number increasing. By examining the SOMO and natural population analysis, we found the excess electron density is localized on the cytidine moiety, especially on the cytosine base unit. This may help explain why the hydrogen bond changes upon the extra electron attachment. In addition, the maps of the reduced density gradient isosurfaces show a rich visualization of the hydrogen bond, van der Waals interaction and steric effect.
 Please wait while we load your content...
Please wait while we load your content...

